Psammaplin A-Modified Novel Radiosensitizers for Human Lung Cancer and Glioblastoma Cells
Article information
Abstract
Background
Psammaplin A (PsA) is a radiosensitizer whereas its clinical application is hampered by poor bioavailability. This study aimed to synthesize novel radiosensitizers using PsA as the lead compound.
Materials and Methods
Eight homodimeric disulfides were synthesized from corresponding acid and cystamine dihydrochloride in N-hydroxysuccinimide and dicyclohexylcarbodiimide coupling conditions. One monomeric thiol analog was obtained by reduction of homodimeric disulfide with dithiothreitol. Clonogenic assay was used to measure cell survival after irradiation and drug treatment in human lung cancer (A549) and glioblastoma (U373MG) cells.
Results and Discussion
Using the PsA backbone, nine compounds were synthesized. Eight compounds showed variable cytotoxicity with 50% inhibitory concentrations ranging 16.14 μM to 150.10 μM (A549), and 13.25 μM to 50.15 μM (U373MG). Four and six compounds radiosensitized A549 and U373MG cells, respectively. Two compounds that radiosensitized both cell lines were tested for its inhibitory effects on DNMT1. One of them was shown to significantly inhibit DNMT1 activity.
Conclusion
Novel compounds with radiosensitizing activity were synthesized. These compounds have a great potential to serve as a basis for the development of future radiosensitizers. Further investigation is warranted for their clinical application.
Introduction
Radiotherapy (RT) is highly effective for cancer treatment, with over 50% of all cancer patients receiving this treatment. The combination of chemotherapeutic agents with RT has significantly improved local control of cancers and patient survival. But some cancers exhibit inherent radiation resistance. The goal of RT is maximizing cell-killing in the cancer cells while minimizing the damage of normal tissues. Studies to improve the therapeutic ratio have resulted in the development of certain compounds that can act to increase the radiosensitivity of cancer cells or to protect the normal cells from RT [1]. Above all, efforts have been made to develop radiosensitizers in order to combine those agents and eventually enhance the efficacy of RT. Not only can radiosensitizers enhance cell lethality at the same RT-doses, it enables to deescalate RT-doses maintaining the cell-killing effects which can subsequently result in reduction of radiation exposure of normal tissues is patients.
DNA methylation is mediated by DNA methyltransferase (DNMT), and it plays an important role in epigenetic regulation of genes leading to suppressed gene expression. Aberrant silencing of tumor suppressor genes is known to be associated with development and progression of cancer [2, 3]. Given that DNMT overexpression has been found in various types of cancers, DNMT inhibitors (DNMTIs) have been investigated as anti-cancer agents [4–6]. DNMTIs are classified into two families, which are nucleosides and non-nucleosides, and various DNMTIs showed anti-cancer activity by themselves or in combination with other cytotoxic agents [7–10]. As a result, drugs such as 5-azacytidine (Azacitidine, Vidaza®) and 5-aza-2′-deoxycytidine (Decitabine, Dacogen®) have actually been approved by the FDA for treatment of myelodysplastic syndrome as well as acute myeloid leukemia [8]. Nucleoside analogues such as 5-azacytidine, zebularine and 5-aza-2′-deoxycytidine enhanced RT lethality in human cancer cell lines [11–14]. Furthermore, DNMTIs are also known to sensitize cancer cells to cell killing by RT. These results suggest the potential application of DNMTIs as radiosensitizers. However, application of nucleoside DNMTIs in cancer treatment has occasionally been restricted due to their toxicity and poor chemical stability [15, 16]. Given the limitations of nucleoside analogues, development of non-nucleoside DNMTIs is needed.
Psammaplin A (PsA) is a symmetrical bromotyrosine-derived disulfide dimer isolated from the marine sponge Pseudoceratina purpurea, and it represents the first example of a disulfide and oxime containing natural product [17]. PsA has been found to have a wide range of bioactivities, especially for antimicrobial and antitumor activity [18]. More to the point, PsA showed an inhibitory activity against both DNMT and histone deacetylase [18–20]. We previously reported that PsA, as a non-nucleoside DNMTI, sensitizes human lung cancer and glioblastoma cells to radiation lethality in vitro [21]. However, PsA is unstable in vivo and has poor bioavailability [22]. To extend or maintain the radiosensitizing activity of PsA towards further studies, we aimed to develop novel PsA-modified derivatives that can serve as future backbones for radiosensitizers. In this study we synthesized and screened the PsA-modified compounds for their radiosensitizing efficacy in human lung cancer and glioblastoma cells.
Materials and Methods
1. Synthesis
Moisture- or air-sensitive reaction was conducted under nitrogen in distilled solvents. The standard reaction work-up involved drying the solution of crude product over anhydrous MgSO4, removing the solvent under reduced pressure. The commercial reagents were purchased from Aldrich, Fluka or Sigma Co. Melting points were measured on Thomas-Hoover melting point apparatus and not corrected. 1H, 13C, HSQC and HMQC NMR spectra were taken on Varian 400 MHz spectrometer in CDCl3, DMSO-d6, or CD3OD. Chemical shifts (δ) are in parts per million (ppm) relative to tetramethylsilane, and coupling constants (J) are in Hertz (Hz). The abbreviations in the NMR spectra are as follows; s: singlet, br: broad, d: doublet, dd: doublet of doublet, t: triplet, quint: quintet, m: multiplet, mp: melting point, ArH: aromatic hydrogen. GC-MS (EI) spectra were obtained on a GCMS QP2010 (Shimadzu Corp., Kyoto, Japan) and Agilent 5975C MSD spectrometer (Agilent, Santa Clara, USA). High resolution fast-atom bombardment mass spectra (HRMS [FAB]) were obtained on a JMS-700 spectrometer (Jeol, Peabody, USA). Medium pressure liquid chromatography (MPLC) was performed on YAMAZEN and fraction collector performed on EYELA DC-1500 (Tokyo Rikakikai Co., Ltd., Tokyo, Japan). An analytical thin layer chromatography (TLC) was performed on pre-coated silica gel 60 F254 plates (Merck & Co., Inc., Kenilworth, USA). Solvent systems for TLC were ethyl acetate/n-hexane and 10% methanol in dichloromethane. Column chromatography was carried out on Merck silica gel 9385 (230–400 mesh) (Merck & Co., Inc.) and eluted with ethyl acetate/n-hexane and methanol/dichloromethane mixture.
1) General procedures for the synthesis of compounds 1–3
To a solution of 4-hydroxyphenylpyruvic acid (900 mg, 5.0 mmol) in absolute ethanol (20 mL), hydroxylamine or O-benzylhydoxylamine hydrochloride (10.0 mmol) and trimethylamine (10.0 mmol) were added and stirred for 5 hours at room temperature. The reaction mixture was rotary evaporated to remove ethanol under reduced pressure. The residue was added with water and acidified with c-HCl and then extracted with ethyl acetate (3·30 mL). The combined organic extract was dried with anhydrous MgSO4, filtrated and concentrated to give crude precipitate which was recrystallized with ethyl acetate and n-hexane mixture to give pure white or pale yellow compound.
(1) 2-(Hydroxyimino)-3-(4-hydroxyphenyl) propanoic acid (1)
Yield: 85%, mp: 156–158°C (lit. 153–155°C [23])
(2) 2-(Benzyloxyimino)-3-(4-hydroxyphenyl) propanoic acid (2)
Yield: 88%, mp: 100–102°C, 1H NMR (DMSO-d6): 9.29 (1H, br s, COOH), 7.28–7.42 (5H, m, ArH), 6.95 (2H, d, J=8.8 Hz, H-2,6), 6.64 (2H, d, J=8.4 Hz, H-3,5), 5.25 (2H, s, CH2), 3.71 (2H, s, CH2); 13C NMR (DMSO-d6): 165.3 (C=O), 154.2 (C=N), 153.1 (C-4), 135.1, 131.8 (C-2,6), 128.6 (C-1), 128.4, 128.1, 116.3 (C-3,5), 76.3 (CH2), 37.6 (CH2), GC-MS (EI) m/z: 285 [M]+.
(3) 2-(Benzyloxyimino)-3-(3-bromo-4-hydroxyphenyl) propanoic acid (3)
Yield: 78%, mp: 149–151°C, 1H NMR (CDCl3): 10.12 (1H, br s, COOH), 7.30–7.37 (5H, m, ArH), 7.26 (1H, d, J=2.0 Hz, H-2), 6.97 (1H, dd, J=8.0, 2.0 Hz, H-6), 6.83 (1H, d, J=8.0 Hz, H-5), 5.26 (2H, s, CH2), 3.71 (2H, s, CH2), 13C NMR (DMSO-d6): 165.9 (C=O), 154.8 (C=N), 151.3 (C-4), 136.3, 132.3, 130.0, 129.5, 128.4, 127.8 (C-1), 127.0, 115.6 (C-5), 110.1 (C-3), 76.5 (CH2), 38.8 (CH2), GC-MS (EI) m/z: 365 [M+1]+, 363 [M-1]+.
2) General procedures for the synthesis of homodimers (4–6 [24], 8, 10, 12)
To a solution of carboxylic acid (1–3, 3,4-difluorocinnamic acid, cinnamic acid, 2-chlorophenylacetic acid, 3-methoxyphenylacetic acid, and benzoic acid, 1.0 mmol) in 1,4-dioxane (20 mL) and dimethylformamide (DMF, 3 mL), N-hydroxysuccinimide (NHS, 1.5 mmol) and dicyclohexylcarbodiimide (DCC, 1.5 mmol) were added at room temperature. After stirring for 1 hour, a mixture of cystamine dihydrochloride (0.5 mmol) and triethylamine (2.0 mmol) in 1,4-dioxane (5 mL) was added at 0°C and stirred for 24 hours at room temperature. After the reaction was over, the reaction mixture was left standing in refrigerator and then filtered. The obtained filtrate was washed with brine and extracted with ethyl acetate (3·30 mL). The extract was dried over anhydrous MgSO4, filtered and concentrated to give a crude compound which was purified by column chromatography (ethyl acetate:n-hexane=1:1 and 2:1) or MPLC to give a pure white or pale yellow compound.
(1) N,N′-Bis(3-(3-bromo-4-hydroxyphenyl)-2-benzyloximinopropionyl)cystamine (4)
Yield: 41%, 1H NMR (CDCl3): 7.32–7.39 (10H, m, ArH), 7.24 (2H, d, J=2.0 Hz, H-2,2′), 7.01 (2H, t, J=6.0 Hz, H-6,6′), 6.85 (2H, d, J=8.0 Hz, H-5,5′), 5.25 (4H, s, 2·CH2), 3.73 (4H, s, 2·CH2), 3.41 (4H, t, J=6.8 Hz, 2·CH2), 2.80 (4H, t, J=6.8 Hz, 2·CH2), 13C NMR (DMSO-d6): 163.1 (C=O), 153.1 (C=N), 150.8 (C-4), 136.3 (ArH), 132.3 (C-2), 130.0 (C-1), 129.5 (C-6), 128.7 (ArH), 128.4 (ArH), 127.6 (ArH), 116.2 (C-5), 110.3 (C-3), 76.2 (CH2), 38.2 (CH2), 37.3 (CH2), 28.8 (CH2), HRMS (FAB): m/z calcd for C36H36Br2N4O6S2 [M+H]+ 843.0521, found 843.0517.
(2) N,N′-Bis(3-(4-hydroxyphenyl)-2-oximinopropionyl) cystamine (5)
Yield: 39%, mp: 160–162°C, 1H NMR (DMSO-d6): 11.73 (2H, s, 2·OH), 9.15 (2H, s, 2·OH), 8.02 (2H, t, J=5.6 Hz, 2·NH), 7.00 (4H, d, J=8.4 Hz, H-2,2′,6,6′), 6.62 (4H, d, J=8.4 Hz, H-3,3′,5,5′), 3.68 (4H, s, 2·CH2), 3.44 (4H, q, J=7.2 Hz, 2·CH2), 2.76 (4H, t, J=7.6 Hz, 2·CH2), 13C NMR (DMSO-d6): 163.1 (C=O), 155.9 (C=N), 151.6 (C-4), 131.3 (C-2,6), 127.8 (C-1), 117.1 (C-3,5), 40.1 (CH2), 38.1 (CH2), 29.8 (CH2), HRMS (FAB): m/z calcd for C22H26N4O6S2 [M+H]+ 507.1372, found 507.1370.
(3) N,N′-Bis(3-(4-hydroxyphenyl)-2-benzyloximinopropionyl) cystamine (6)
Yield: 38%, 1H NMR (DMSO-d6): 7.29–7.38 (10H, m, ArH), 7.14 (4H, d, J=10.0 Hz, H-2,2′,6,6′), 6.69 (4H, d, J=9.6 Hz, H-3,3′,5,5′), 5.19 (4H, s, 2·CH2), 3.89 (4H, s, 2·CH2), 3.51 (4H, q, J=7.6 Hz, 2·CH2), 2.69 (4H, t, J=8.8 Hz, 2·CH2), 13C NMR (DMSO-d6): 165.2 (C=O), 155.1 (C=N), 154.5 (C-4), 135.2 (ArH), 132.2 (C-2,6), 128.2 (C-1), 128.7 (ArH), 128.5 (ArH), 127.8 (ArH), 116.3 (C-3,5), 76.2 (CH2), 39.1 (CH2), 38.2 (CH2), 29.9 (CH2), HRMS (FAB): m/z calcd for C36H38N4O6S2 [M+H]+ 687.2311, found 687.2307.
(4) N,N′-Bis(3,4-Difluorocinnamoyl) cystamine (8)
Yield: 73%, mp: 160–162°C, 1H NMR (DMSO-d6): 8.33 (2H, t, J=5.6 Hz, NH), 7.63 (2H, dd, J=5.6, 8.4 Hz, H-6,6′), 7.36–7.48 (6H, m, 2·CH, H-2,2′,5,5′), 6.62 (2H, d, J=16.0 Hz, 2·CHCO), 3.48 (4H, q, J=6.4 Hz, 2·CH2), 2.86 (4H, t, J=6.8 Hz, 2·CH2), 13C NMR (DMSO-d6): 164.6 (CO), 149.9 (C-3, dd, J=12.0, 246.2 Hz), 149.7 (C-4, dd, J=12.0, 239.0 Hz), 136.7 (=CH), 132.7 (C-1, t, J=4.2 Hz), 124.6 (C-6, dd, J=2.0, 17.6 Hz), 123.2 (=CHCO), 117.8 (C-5, dd, J=4.0, 17.8 Hz), 116.1 (C-2, J=17.9 Hz), 38.0 (CH2), 37.1 (CH2), HRMS (FAB): m/z calcd for C22H20BrF4N2 O2S2 [M+H]+ 485.0981, found 485.0978.
(5) N,N′-Bis(cinnamoyl)cystamine (9)
Yield: 78%, mp: 121–123°C, 1H NMR (DMSO-d6): 8.35 (2H, t, J=5.6 Hz, 2·NH), 7.55 (4H, d, J=6.8, Hz, H-2,2′,6,6′), 7.44 (2H, d, J=16.0 Hz, 2·CH), 7.34–7.42 (6H, m, H-3,3′,4,4′,5,5′), 6.64 (2H, d, J=16.0 Hz, 2·CHCO), 3.50 (4H, q, J=6.4 Hz, 2· CH2), 2.88 (4H, t, J=6.4 Hz, 2·CH2), 13C NMR (DMSO-d6): 165.8 (CO), 139.6 (=CH), 135.5 (C-1), 130.1 (C-4), 129.6 (C-2,6), 128.2 (C-3,5), 122.6 (=CHCO), 38.8 (CH2), 38.0 (CH2), HRMS (FAB): m/z calcd for C22H24N2O2S2 [M+H]+ 413.1357, found 413.1353.
(6) N,N′-Bis(2-(2-chlorophenyl)acetyl)cystamine (10)
Yield: 74%, 1H NMR (DMSO-d6): 8.24 (2H, t, J=5.2 Hz, 2·NH), 7.40 (2H, dd, J=2.4, 6.6 Hz, H-3,3′), 7.34 (2H, dd, J=2.0, 6.4 Hz, H-6,6′), 7.22–7.32 (4H, m, H-4,4′,5,5′), 3.57 (4H, s, 2·CH2), 3.56 (4H, q, J=6.0 Hz, 2·CH2), 2.79 (4H, t, J=6.8 Hz, 2·CH2); 13C NMR (DMSO-d6): 170.5 (C=O), 134.3 (C-1), 134.2 (C-6), 132.6 (C-2), 129.7 (C-3), 129.3 (C-4), 127.8 (C-5), 40.5 (CH2), 38.6 (CH2), 37.6 (CH2), HRMS (FAB): m/z calcd for C20H22Cl2N2O2 S2 [M+H]+ 457.0578, found 457.0575.
(7) N,N′-Bis(2-(3-methoxyphenyl)acetyl)cystamine (11)
Yield: 81%, mp: 108–110°C, 1H NMR (DMSO-d6): 8.22 (2H, t, J=5.2 Hz, 2·NH), 7.19 (2H, t, J=7.6 Hz, H-5,5′), 6.76–6.85 (6H, m, H-2,2′,4,4′,6,6′), 7.22–7.32 (4H, m, H-4,4′,5,5′), 3.72 (6H, s, 2·CH3), 3.38 (4H, s, 2·CH2), 3.32 (4H, q, J=6.4 Hz, 2·CH2), 2.76 (4H, t, J=6.4 Hz, 2·CH2), 13C NMR (DMSO-d6): 171.4 (C=O), 159.8 (C-3), 138.2 (C-1), 130.0 (C-5), 121.9 (C-6), 115.3 (C-2), 112.5 (C-4), 55.6 (OCH3), 43.0 (CH2), 38.6 (CH2), 37.7 (CH2), HRMS (FAB): m/z calcd for C22H26N2O4S2 [M+H]+ 448.1490, found 448.1487.
(8) N,N′-Dibenzoylcystamine (12)
Yield: 80%, mp: 132–134°C (lit. 130–132°C [25]), 1H NMR (DMSO-d6): 8.65 (2H, s, 2·NH), 7.83 (4H, d, J=7.6 Hz, H-2,2′, 6,6′), 7.40–7.60 (6H, m, H-3,3′,4,4′,5,5′), 3.56 (4H, q, J=6.0 Hz, 2·CH2), 2.93 (4H, t, J=6.4 Hz, 2·CH2), 13C NMR (DMSO-d6): 168.0 (C=O), 134.5 (C-1), 132.3 (C-4), 129.2 (C-3,5), 127.7 (C-2,6), 38.7 (CH2), 37.7 (CH2), HRMS (FAB): m/z calcd for C18H20N2O2S2 [M+H]+ 361.1044, found 361.1040.
(9) N-Cinnamoylcysteamine (13)
To a solution of N,N′-bis(cinnamoyl)cystamine (9, 412 mg, 1 mmol) in methanol (30 mL) and dimethylformamide (5 mL), 1 M KOH (0.1 mL) and dithiothreitol (462 mg, 3 mmol) were added at room temperature. The reaction mixture was quenched with 0.5 M HCl at 0°C after 1 hour and extracted with dichloromethane (3·30 mL). The combined extracts were washed with water and brine, and were dried over anhydrous MgSO4, and then filtered. The filtrate was evaporated to give crude oily compound. The desired compound 13 was obtained as a colorless oil by short column chromatography (ethyl acetate:n-hexane=1:1). Yield: 88%, 1H NMR (DMSO-d6): 8.33 (1H, t, J=5.6 Hz, NH), 7.53 (1H, d, J=6.8 Hz, H-2,6), 7.42 (1H, d, J=16.0 Hz, =CH), 7.35–7.43 (3H, m, H-3, 4,5), 6.63 (1H, d, J=16.0 Hz, =CHCO), 3.48 (2H, q, J=6.8 Hz, CH2), 2.79 (2H, q, J=6.4 Hz, CH2), 2.43 (1H, t, J=6.8 Hz, SH), 13C NMR (DMSO-d6): 164.9 (C=O), 139.2 (=CH), 134.8 (C-1), 129.2 (C-2,6), 127.9 (C-3,5), 122.0 (=CHCO), 38.5 (CH2), 37.5 (CH2), HRMS (FAB): m/z calcd for C11H13NOS [M+H]+ 208. 0796, found 208.0792.
2. Cell Culture
A549 (human lung cancer) and U373MG (human glioblastoma) cell lines were purchased from the Korean Cell Line Bank. Cells were cultured and maintained in RPMI media (Welgene, Daegu, Korea), supplemented with 10% fetal bovine serum and 12.5 μg·mL−1 of gentamicin. All compounds were dissolved in DMSO as concentrated stock solutions at 20 mM concentration, corresponding to concentrations of 2.66, 3.37, 2.02, 1.94, 1.65, 1.83, 1.79, 1.44, and 0.83 mg/0.2 mL for PsA, 4, 5, and 8–13, respectively. They were stored at −20°C, and diluted at the time of use.
3. Clonogenic assay
Cell survival after treatment with PsA-modified derivatives and/or irradiation was measured using clonogenic assay as previously reported [21, 22]. Briefly, cells were trypsinized, and appropriate number of cells were seeded into 6 well plates and incubated for 24 hours. Then, cells were treated with nine synthesized compounds or DMSO (for the drug-free groups) at specified concentrations for additional 24 hours. After specified drug treatment, the media was replaced with fresh drug-free medium. Then, to determine the 50% inhibitory concentrations (IC50) of each compound, cells were incubated for another 12 days to allow for colony formation. To screen for the radiosensitizing properties of each compound, cells were irradiated using 6 MV X-ray from a linear accelerator (Clinac 6EX or Clinac 21EX, Varian Medical systems, Palo Alto, USA) at a dose of 0, 2, 4, 6, and 8 Gy immediately after the media was changed (post-seeding 48 hours), and then also incubated for 12 additional days. Then, colonies were fixed with methanol, stained with 0.5% crystal violet, and counted. Only colonies containing 50 cells or more were counted.
4. DNMT1 inhibition assay
Assaying DNMT1 inhibitory activity of compounds was done using the colorimetric EpiQuik DNMT activity/inhibition assay Ultra kit (Epigentek, Farmingdale, USA) according to the manufacturer’s instructions. Briefly, cells were treated with compounds 5 and 10 at a concentration of 100 μM for 24 hours, and DNA was extracted from cells. Then, the colorimetric assay for methylated DNA was performed in 96-well plates. A microplate reader with a 450-nm filter read each plate. The DNMT1 activity was proportional to sample optical density (OD). DNMT1 inhibition was calculated as follows: DNMT1 inhibition (%)=[1-(inhibitor sample OD-blank OD)]·100/(no inhibitor sample OD-blank OD). All data were obtained from triplicate experiments.
5. Statistical analysis
Cell survival data were presented as means of surviving fractions from triplicate experiments. Kaleidagraph version 3.51 (Synergy Software, Reading, USA) was used to fit surviving fractions of irradiated cells to a linear quadratic model of radiation cell killing. Parameters of fitted linear quadratic models were used to calculate radiation doses to obtain specified surviving fractions. Sensitizer enhancement ratio (SER) at surviving fraction of 0.2 was calculated as previously reported [21, 22]. To determine whether the required radiation dose for surviving fraction of 0.2 was significantly lower in the group treated with both PsA-modified derivative and radiation compared to the group which received radiation only from triplicate experiments of a PsA derivative, the ratio paired t test was performed. The level of statistical significance was set at p<0.05. Statistical Package for the Social Sciences version 22.0 (IBM SPSS, Armonk, USA) was used for statistical testing.
Results and Discussion
1. Synthesis
Using PsA as a lead compound, nine PsA-modified compounds were synthesized. They are eight homodimers (4–6 and 8–12) and one monomer (13). As shown in scheme 1, 4-hydroxyphenylpyruvic acid was reacted with hydroxylamine and O-benzylhydroxylamine to afford oxime 1 and O-benzyloxime 2, respectively. Compound 2 was brominated to form bromotyrosine analog 3 by KBr and KBrO3 in an acidic condition [26]. Compound 3 was reacted with cystamine dihydrochloride and triethylamine to synthesize bromotyrosine-derived disulfide analog 4 by N-hydroxysuccinimide (NHS) and dicyclohexylcarbodiimide (DCC) coupling reaction. The synthesis of homodimeric tyrosine-derived disulfide analog 5 and 6, which have no 3-bromo group in their structures, were achieved by amide coupling of compound 1 and 2 with cystamine dihydrochloride using NHS and DCC.
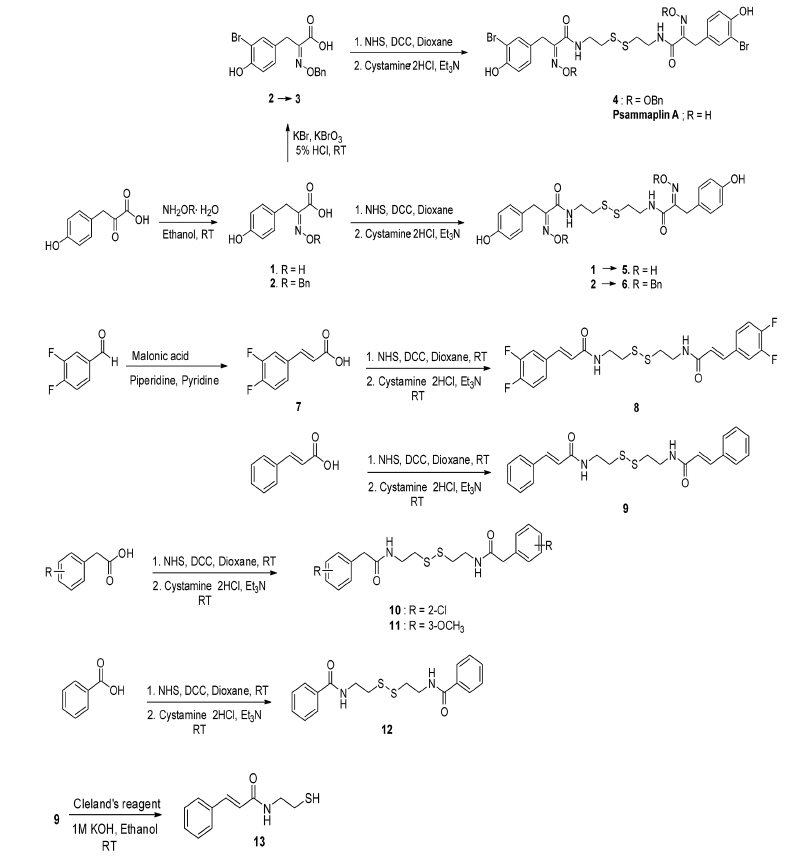
Synthesis of homodimeric disulfides (4–6 and 8–12) and a monomer (13).
Apart from tyrosine-derived disulfide analogs, 3,4-difluorocinnamic acid (7), cinnamic acid, 2-chlorophenylacetic acid, 3-methoxyphenylacetic acid, and benzoic acid were coupled with cystamine dihydrochloride to afford four homodimeric disulfide analogs, 8–12 in moderate yields, respectively. Compound 7 was obtained from 3,4-difluorobenzaldehyde and malonic acid in the presence of piperidine and pyridine. N,N′-Bis(cinnamoyl)cystamine (9) was reduced by dithiothreitol (Cleland’s reagent) to afford N-cinnamoylcysteamine (13) as monomer.
2. Inhibition of cell survival
Clonogenic cell survivals for PsA and the nine synthesized compounds (4–6 and 8–13) were measured, and all compounds reduced the viability of cells in a dose-dependent manner. The IC50 values of compounds are shown in Table 1.

Cytotoxicity of PsA and Its Derivatives
3. Screening of the compounds as radiosensitizers
Clonogenic cell survival was measured after combination of X-ray RT and treatment of seven compounds (4, 5, 8–11, and 13) at measured respective IC50s for 24 hours prior to X-ray RT. However, since compound 5, 8, 9, and 11 were not shown to radiosensitize A549 and U373MG cells at the concentration of around each IC50 value, concentrations of the compounds were subsequently increased (concentration displayed on Figure 1). Radiation survival curves were compared between cells treated with synthesized compounds and those treated with DMSO as the control (Figure 1). The surviving fractions of each compound were measured in a range 0–8 Gy and shown as the mean±SD from triplicate experiments.
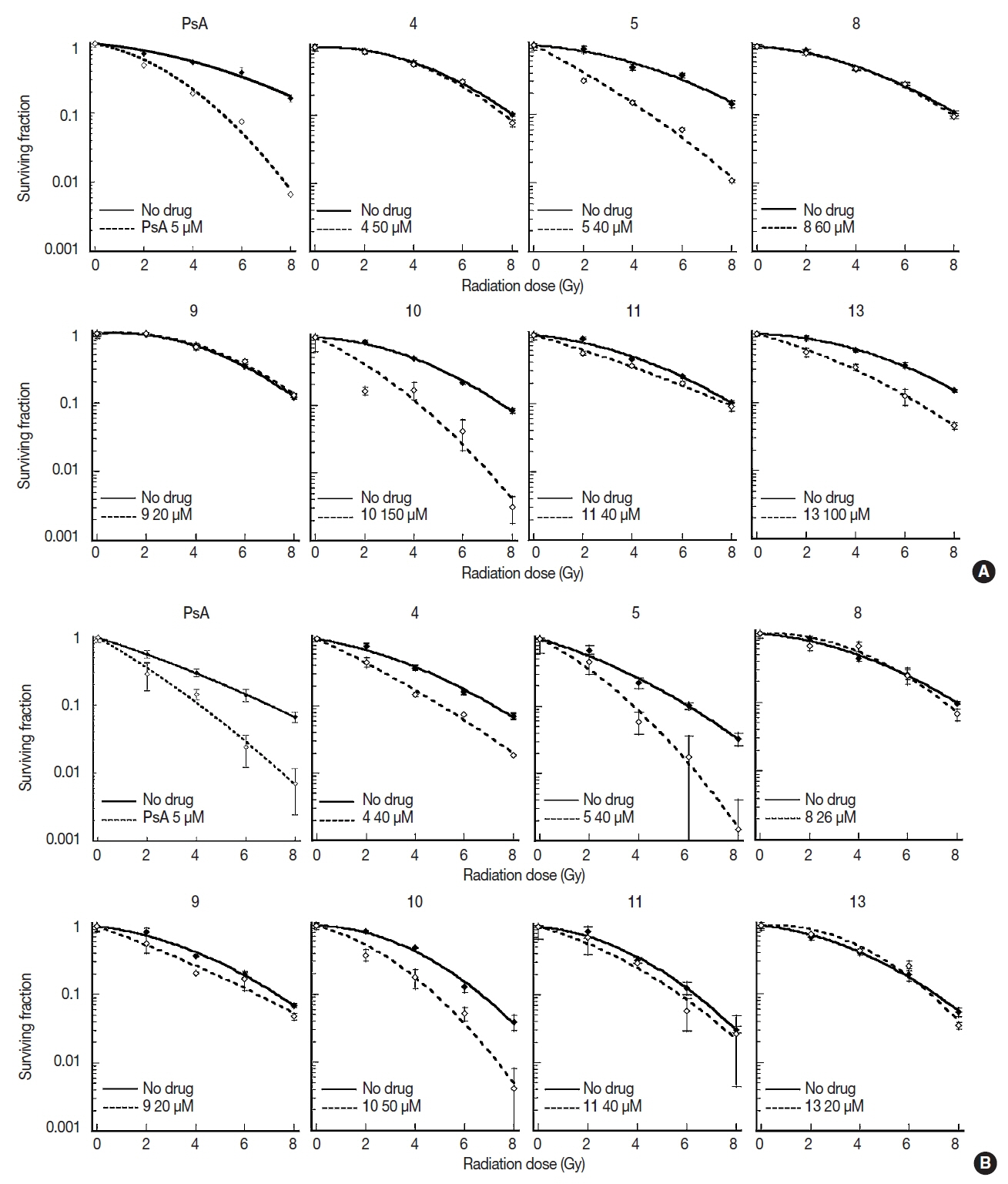
Clonogenic cell survival in (A) A549 and (B) U373MG cells after combination of irradiation and treatment of synthesized compounds. Cells were treated with compounds for 24 hours before irradiation to graded doses of X-ray. Surviving fractions are shown mean±SD from triplicate experiments. PsA, psammaplin A.
The SERs are shown in Figure 2. Two compounds (10 and 13) significantly (p<0.05), and two other compounds (5 and 8) marginally (p<0.10) enhanced cell killing by RT in A549 cells. Five compounds (4, 5, and 9–11) induced significant (p<0.05) radiosensitization whereas one compound (8) demonstrated a marginal (p<0.10) radiosensitization in U373MG cells. Compounds 5, 8, and 10 showed at least marginal radiosensitization in both cell lines. However, although statistically marginal, 8 minimally radiosensitized A549 cells with a mean SER value of 1.08. Thus, only 5 and 10 were selected for DNMT1 inhibition assay.
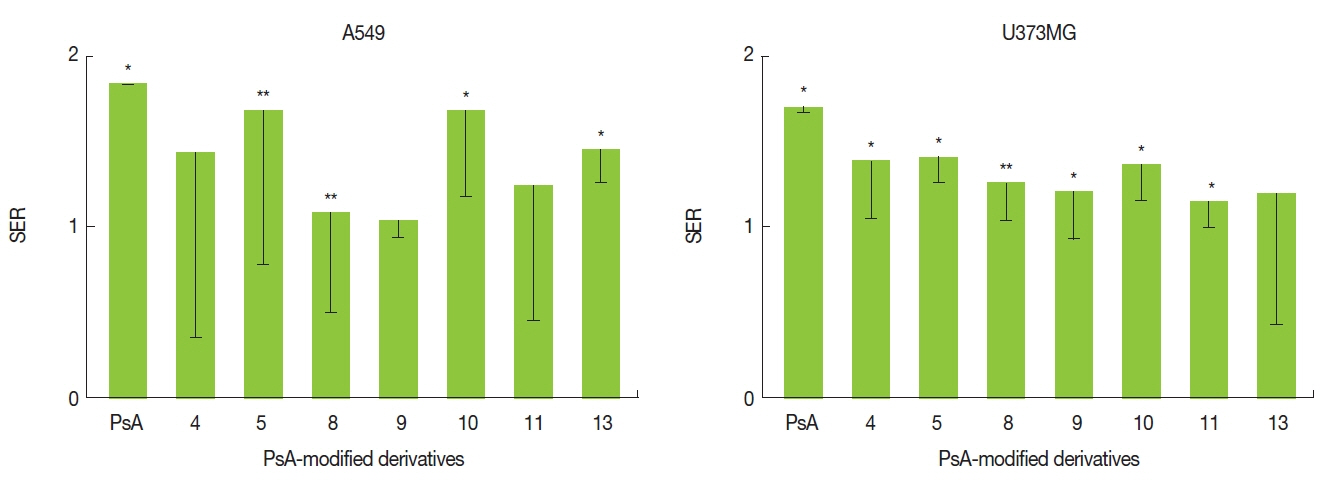
Sensitizer enhancement ratio (SER) of synthetized compounds. SER is a ratio of radiation doses necessary in the absence of each compound compared to in the presence of each compound to produce a surviving fraction of 0.2. SERs are shown as mean with lower limits of the 95% confidence intervals (n=3). *p<0.05, **p<0.1 by ratio paired t test. PsA, psammaplin A.
4. Inhibition of DNMT1 activity
DNMT1 activity was significantly inhibited by PsA and compound 5 at 100 μM, with inhibition rates of 99.99% and 90.57%, respectively. However, DNMT activity was not affected by treatment with 10 (Figure 3).
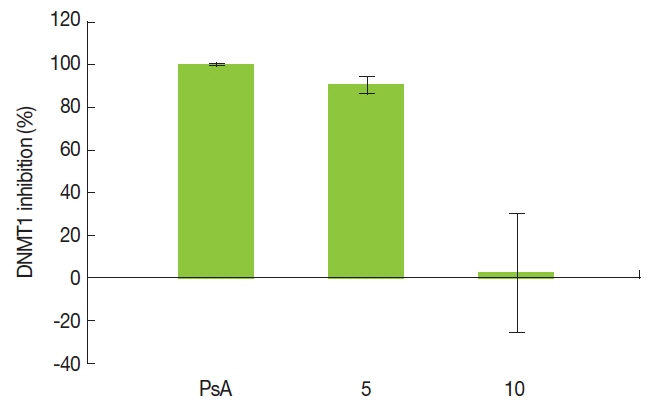
Inhibition of DNMT1 activity by psammaplin A (PsA), 5 and 10 when treated at a concentration of 100 μM for 24 hours. DNMT1 activity was assayed using a DNMT activity assay kit. All data are shown as mean±SD (n=3).
Discussion
The therapeutic ratio of RT can be improved by chemical agents that sensitize cancer cell to the cytotoxic effects of ionizing radiation. We have previously reported that PsA is a potent radiosensitizer in vitro with poor bioavailability in vivo [21, 22]. The present study aimed to synthesize novel compounds that retain radiosensitizing properties of PsA. Such strategy has successfully generated a novel histone deacetylase inhibitor with both in vivo radiosensitizing capacity and biostability [27, 28].
Lee et al. [29] reported that 4-hydroxy modified (4-benzyloxy, 4-tert-butyl, 4-ethoxy-) derivatives of PsA which have no 3-bromo group in their structure were synthesized and demonstrated improved pharmacokinetic properties. In our study, since the oxime is known as an unstable group, one chemical approach to improve the bioavailability was to mask it by protection. We protected the oxime unit of PsA with benzyl group to afford O-benzyl oxime analog 4. For further study on how the bromo group of PsA influenced radiosensitivity, oxime analog 5 and O-benzyl oxime analog 6, which have no 3-bromo groups were synthesized.
There is a limitation in commercial availability of tyrosine and phenylpyruvic acid to synthesize the derivatives of PsA with diverse aromatic substitution patterns. Structurally, the PsA derivatives are composed of a cap structure and a linker which can prepare the homodimer, monomer, and heterodimer. We adopted a commercially available carboxylic acid containing aromatic ring instead of tyrosine moiety as a cap substructure, which would allow better physicochemical properties. Fluoro group may increase a target compound’s lipophilicity and is considered importantly when obtaining molecules that are designed to be active in vivo. Fluorine can also aid hydrophobic interactions between the drug and binding sites on receptors or enzymes [30]. The chlorine substituent is known as a more lipophilic group than a fluoro- or methoxy- substituent, so it was selected for improving the bioavailability. 3,4-Difluorocinnamic acid, cinnamic acid, 2-chlorophenylacetic acid, 3-methoxyphenylacetic acid and benzoic acid, instead of the tyrosine part (4-hydroxyphenylpyruvic acid), were used for the synthesis of five homodimeric disulfide derivatives (8–12).
As shown in Figures 2 and 3, compounds 5, 8, 10, and 13 radiosensitized the A549 cells, and 4, 5, and 9–11 radiosensitized the U373MG cells, in vitro. Especially, N,N′-bis(3-(4-hydroxyhenyl)-2-oximinopropionyl)cystamine (5) and N,N′-bis(2-(2-chlorophenyl)acetyl)cystamine (10) enhanced the radiation lethality by around 1.5 times in both cell lines. Both 5 and 10 are of a homodimeric disulfide structure, and are potent radiosensitizers comparable to PsA (Figure 2). However, O-benzyl oxime analog 4 did not demonstrate a statistically significant radiosensitizing effect in A549 cells. The benzyl protection of oxime seemed to result in reduced radiosensitization. Future studies should be performed whether the radiosensitizing effects of the compounds are affected by drug concentration.
Along with histone deacetylation, DNA methylation is a major mechanism of epigenetic alteration. Thus, inhibitors of epigenetic modulators such as histone deacetylase and DNMT have been investigated for anti-tumor activity in clinical trials. Inhibition of DNMT as well as histone deacetylase has been reported to sensitize human cancer cells to radiation-induced cell killing [21, 22]. However, no epigenetic modulators are currently used in conjunction with RT in clinical settings partly due to biochemical instability.
We have previously reported that a possible underlying mechanism of radiosensitization by a known DNMTI, PsA, is via inhibiting the DNA damage repair process [21]. Since 5 and 10 are PsA-modified homodimeric disulfides, we evaluated the inhibition effect on DNMT1 activity in 2 compounds at a concentration of 100 μM. As a result, 5 showed DNMT1 inhibitory activity comparable to PsA. However, it is remarkable that, 10 did not show any inhibition of DNMT1 (Figure 3). It is unknown whether DNMT inhibitory activity is prerequisite of radiosensitization by PsA. Despite the absence of DNMT1 inhibitory activity, 10 was as a potent a radiosensitizer. Thus, our observations may suggest that compound 10 confers radiosensitizing properties regardless of DNMT1 inhibitory activity. However, since the concentration of compounds used for the DNMT inhibition assay was very high, these results of DNMT inhibition assay may not have a biological meaning. Furthermore, since no mechanistic investigation was performed, it is to be elucidated whether 10 inhibits different isotypes of DNMTs.
From this result, we fortunately found that the thiol monomers, the major fragment of disulfide derivatives, are similar with synthetic thiol compounds which are known as radiopropectors. Of these series, amifostine (WR-2721, Ethyol®, 2-(3-amniopropyl)ethylsulfanylphosphonic acid) is the only radioprotector that has been clinically approved by the FDA. An inactive prodrug, amifostine, is transformed to an active free thiol (WR-1065) by dephosphorylation by alkaline phosphatase in normal tissues. Generally, normal tissues are able to contain free thiol up to a concentration 100-fold higher than tumors and thus can the free thiols protect normal tissues selectively [31]. Like all other drugs, amifostine has side effects such as acute hypotension, severe nausea, vomiting and allergy when administered intravenously [32]. In the future, PsA-modified derivatives will be studied for the potential dual effects of radiosensitization and radioprotection.
Conclusion
Using PsA as a lead structure, we synthesized and identified nine compounds that showed potent in vitro radiosensitization in two human cancer cell lines, lung cancer and glioblastoma. The present study suggests that the synthesized compounds have a great potential to serve as structural backbones for development of novel radiosensitizers further clinical application. Future investigations are warranted to confirm the bioavailability, radiosensitizing properties in vivo, mechanism, and safety of the compounds.
Acknowledgements
Funding: This study was supported by the Korean government (MSIP) through the National Research Foundation of Korea grant 2013M2A2A7043683 (Il Han Kim), and the Seoul National University Hospital through SNUH CRI 0320170080 (Il Han Kim) and SNUH CRI 0320130400 (Hak Jae Kim). Authors thank Eun Jung Oh for her administrative and management support of the research project.